SACHIN SHANBHAG |


 Subramanian, G.; Shanbhag, S.; "Conformational properties of blends of cyclic and linear polymer melts", Phys. Rev. E, 2008, 77(1), 011801.
Subramanian, G.; Shanbhag, S.; "Conformational properties of blends of cyclic and linear polymer melts", Phys. Rev. E, 2008, 77(1), 011801.
Using an adapted annealing algorithm to identify primitive paths of a melt of ring polymers in ring-linear blends, we found that the primitive path length and the average number of entanglements of the linear component were independent of the blend composition. However, the the same quantities on a ring molecule increased approximately linearly with the fraction of linear chains, and for large N, they approached values comparable with linear chains. Threading of ring molecules by linear chains and we conjectured that for large N, these latter interactions facilitate the formation of a percolating entangled network, thereby resulting in a disproportionate retardation of the dynamical processes. Online article |
 Shanbhag, S.; Alamo, R.G., "On the thermodynamic driving force for nucleation at large undercoolings",
Polymer, 2008, 49, 2515.
Shanbhag, S.; Alamo, R.G., "On the thermodynamic driving force for nucleation at large undercoolings",
Polymer, 2008, 49, 2515.
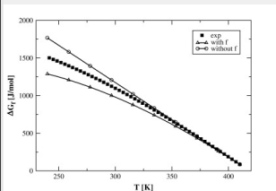
The molar free energy difference between the amorphous and crystalline phases is an important quantity in studying both the thermodynamics and kinetics of polymers. In this work we computed the free energy using heat capacity data over a large temperature range between the melting and the glass transition temperatures. We used these values to assess the validity of the approximate correction factors first proposed by Hoffman, and widely (and somewhat indiscriminately!) used in the literature for a variety of polymers to account for the change in free energy with temperature. Surprisingly, for polyethylene and isotactic polypropylene, which are industrially important polymers, it was found that the performance of these correction factors worsened uncorrected estimates. Online article |
 Iyer, B.V.S.;Shanbhag, S.; Juvekar, V.A.; Lele, A.K., "Self-diffusion coefficient of ring polymers in semidilute solution",
J. Polym. Sci.: Polym. Phys., 2008, 46(21), 2370.
Iyer, B.V.S.;Shanbhag, S.; Juvekar, V.A.; Lele, A.K., "Self-diffusion coefficient of ring polymers in semidilute solution",
J. Polym. Sci.: Polym. Phys., 2008, 46(21), 2370.
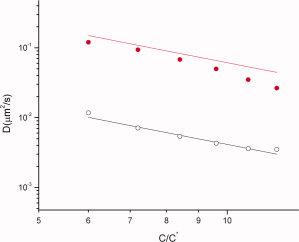
In a topologically constraining environment the size of a flexible nonconcatenated ring polymer and its dynamics are known to differ from that of linear polymers. Hence, the diffusion coefficient of ring polymers can be expected to be different from linear chains. Here, we present scaling arguments for the concentration and molecular weight dependence of self-diffusion coefficient of ring polymers in semidilute solutions, and show that contrary to expectations these scaling relations are identical to what is known for linear polymers. At higher concentrations excluded volume interactions arising from possibilities of segmental overlap can become effective for large ring polymers. In this regime the diffusion coefficient of large ring polymers shows a relatively weaker dependence on concentration and molecular weight. Online article |
 Subramanian, G.; Shanbhag, S.; "Self-Diffusion in Binary Blends of Cyclic and Linear Polymers", Macromolecules, 2008, 41(19), 7232.
Subramanian, G.; Shanbhag, S.; "Self-Diffusion in Binary Blends of Cyclic and Linear Polymers", Macromolecules, 2008, 41(19), 7232.
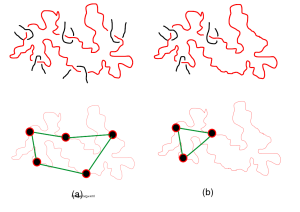
In this paper, we used and extended our lattice simulations to estimate the self-diffusivity of entangled cyclic and linear polymers in blends of varying compositions. To interpret simulation results, we proposed a minimal constraint release model for the motion of a cyclic polymer infiltrated by neighboring linear chains. Both the simulation and recently reported experimental data on entangled DNA solutions support the simple model over a wide range of blend compositions, concentrations, and molecular weights. Online article |
 Subramanian, G.; Shanbhag, S.; "On the relationship between two popular lattice models for polymer melts", J. Chem. Phys., 2008, 129, 144904.
Subramanian, G.; Shanbhag, S.; "On the relationship between two popular lattice models for polymer melts", J. Chem. Phys., 2008, 129, 144904.
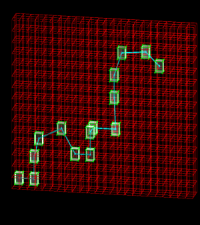
In this paper, we tried to a figure out whether we could establish a mapping between two successful lattice bond-fluctuation models for polymers [I. Carmesin and K. Kremer, Macromolecules 21, 2819 (1988); J. S. Shaffer, J. Chem. Phys. 101, 4205 (1994)] which taken together have been used in about 200 simulation studies. Simulations conducted using both models, and previously published data were compared in an attempt to establish relationships between molecular weight, lengthscale, and timescale. Using these relationships, an examination of the self-diffusion coefficient yields excellent agreement not only between the two models, but also with experimental data on polystyrene, polybutadiene, and polydimethylsiloxane. However, it is shown that even with the limited set of criteria examined in this a true mapping between these two models is elusive. Nevertheless, a practical guide to convert between models is provided. Online article |
 Qin, B.; Zhao, Z.; Song, R.; Shanbhag, S.; Tang, Z.,"A Temperature-Driven Reversible Phase Transfer of 2-(Diethylamino)ethanethiol-Stabilized CdTe Nanoparticles",
Angew. Chemie , 2008, 47(51), 9875.
Qin, B.; Zhao, Z.; Song, R.; Shanbhag, S.; Tang, Z.,"A Temperature-Driven Reversible Phase Transfer of 2-(Diethylamino)ethanethiol-Stabilized CdTe Nanoparticles",
Angew. Chemie , 2008, 47(51), 9875.
By changing the temperature, we found that DEAET-stabilized CdTe nanoparticles could transfer reversibly from the aqueous phase to the organic phase, which opens up a whole new set of applications in which the nanoparticles could be used to transport material between organic and aqueous phases. Theoretically, and experimentally, we found that DEAET molecules, by themselves show no such phase transfer. Compared to DEAET molecules, the different phase transfer behavior of CdTe NPs is ascribed to their size effects, which are large enough to disturb the hydrogen bonding of water surrounding NPs and thus increase the solubility in water. Online article |
Back to Top
© 2008 Sachin Shanbhag
Last Modified: 10/01/2008
Last Modified: 10/01/2008